The Atlas pipeline¶
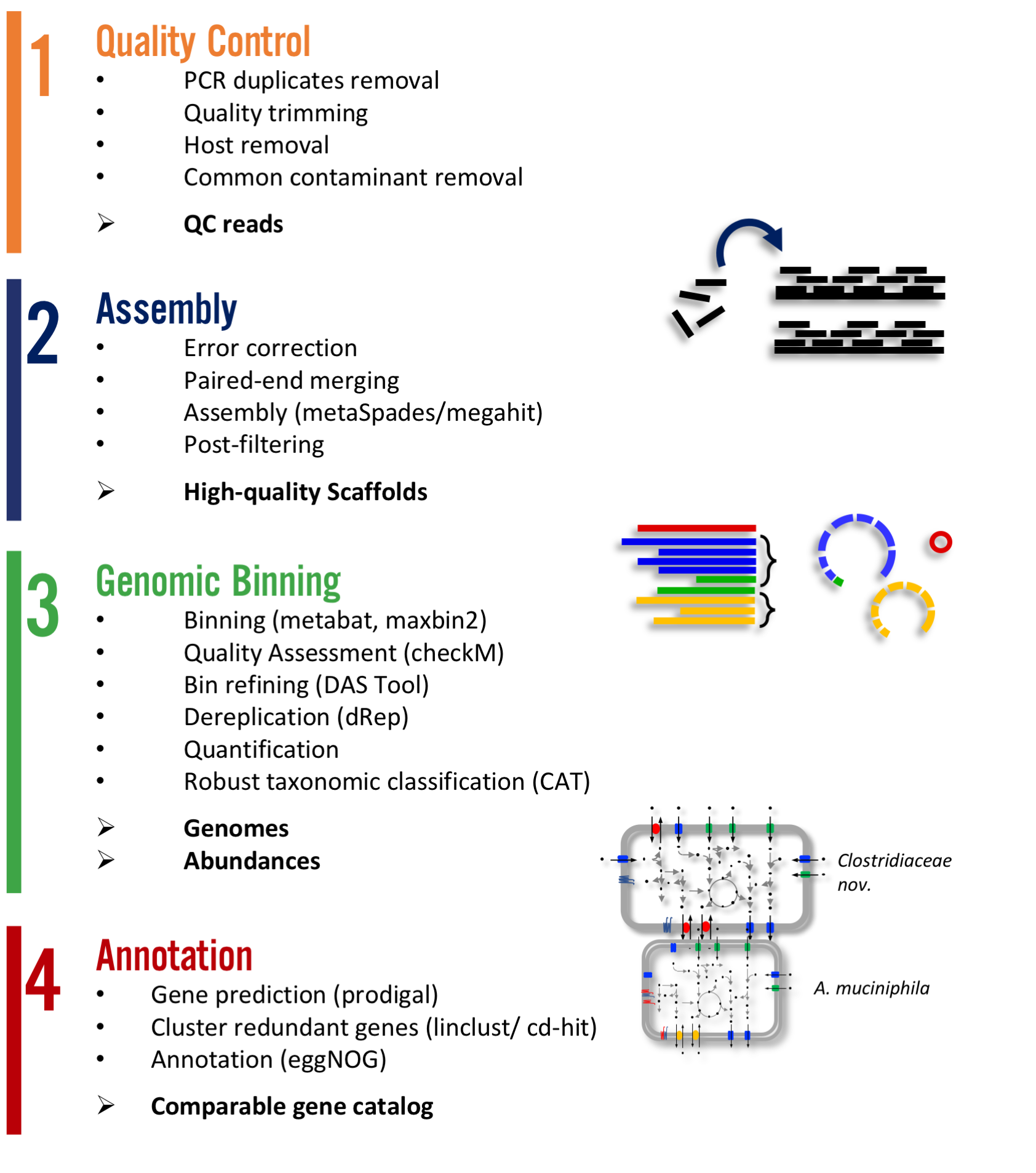
Expected output¶
Quality control¶
atlas run qc
#or
atlas run all
Runs quality control of single or paired end reads and summarizes the main QC stats in reports/QC_report.html.
Per sample it generates:
{sample}/sequence_quality_control/{sample}_QC_{fraction}.fastq.gz- Various quality stats in {sample}/sequence_quality_control/read_stats
Fractions:¶
When the input was paired end, we will put out three the reads in three fractions R1,R2 and se The se are the paired end reads which lost their mate during the filtering. The se are seamlessly integrated in the next steps.
Assembly¶
atlas run assembly
#or
atlas run all
Besides the reports/assembly_report.html this rule outputs the following files per sample:
{sample}/{sample}_contigs.fasta{sample}/sequence_alignment/{sample}.bam{sample}/assembly/contig_stats/postfilter_coverage_stats.txt{sample}/assembly/contig_stats/prefilter_contig_stats.txt{sample}/assembly/contig_stats/final_contig_stats.txt
Genomes¶
atlas run genomes
#or
atlas run all
Binning¶
When you use different binners (e.g. metabat, maxbin) and a binner-reconciliator (e.g. DAS Tool), then Atlas will produce for each binner and sample:
{sample}/binning/{binner}/cluster_attribution.tsv
which shows the attribution of contigs to bins. For the final_binner it produces the
reports/bin_report_{binner}.html
See an example
As a summary of the quality of all bins. These bins are then De-replicated using DeRep. The Metagenome assembled genomes are then renamed, but we keep mapping files.
genomes/Dereplicationgenomes/clustering/contig2genome.tsvgenomes/clustering/allbins2genome.tsv
The main output files¶
genomes/genomesgenomes/annotations/genesgenomes/checkm/completeness.tsvgenomes/taxonomy/taxonomy_names.tsvgenomes/counts/median_coverage_genomes.tsvgenomes/counts/raw_counts_genomes.tsv
Gene Catalog¶
atlas run all
# or
atlas run genecatalog
The gene catalog takes all genes predicted from the genomes and clusters them according to the configuration. This rule produces the following output file for the whole dataset.
Genecatalog/gene_catalog.fnaGenecatalog/gene_catalog.faaGenecatalog/annotations/eggNog.tsv